post process energy calculation

Hello, sfepy group,
I have a question about post-process calculations. For my problem I used the modified examples linear_elastic.py and hyperelastic.py. After the calculation of strain and stress tensors I need to get energy of deformation
\int_{V} S : C, where C = 2*E+I.
I think I need to calculate the multiplication of tensors first, as some material variable, and then integrate it. So I have 2 questions:
How can I calculate the double scalar multiplication of two arrays strain and stress that have shape (number of elements, 1, 6, 1)? (Double scalar multiplication of A and B is a scalar that can be got as A_{ij}B_{ji}). Because of the shape of arrays I can't use numpy.inner(stress,strain). I made some function for these calculation, but perhaps, the simplier way exsists.
Then I get the multiplication - the scalar function of energy I should integrate it. For this I get the dw_volume_integrate term. I'm trying to integrate material parameter, where energy has been saved.
solid.update({ 'E' : get_W(stress,strain),#energy in elements }) U = problem.evaluate('dw_volume_integrate.i1.Omega(solid.E, v)',)
Or may be I should use some new variable and initialize it in post-proc. I'm asking for help in correct formulation of this integral. Should I define energy as material parameter or some new variable? And how I can do it.
Thanks in advance,

Hi Anastasia,
I am going to EuroSciPy today, so I will answer in detail after/if I get the connectivity there.
Short answer now: look at sfepy.linalg.utils.dot_sequences() or numpy.einsum() for generalized dot products.
r.
On 08/19/2013 12:51 PM, Anastasia Konovalova wrote:
Hello, sfepy group,
I have a question about post-process calculations. For my problem I used the modified examples linear_elastic.py and hyperelastic.py. After the calculation of strain and stress tensors I need to get energy of deformation
\int_{V} S : C, where C = 2*E+I.
I think I need to calculate the multiplication of tensors first, as some material variable, and then integrate it. So I have 2 questions:
How can I calculate the double scalar multiplication of two arrays strain and stress that have shape (number of elements, 1, 6, 1)? (Double scalar multiplication of A and B is a scalar that can be got as A_{ij}B_{ji}). Because of the shape of arrays I can't use numpy.inner(stress,strain). I made some function for these calculation, but perhaps, the simplier way exsists.
Then I get the multiplication - the scalar function of energy I should integrate it. For this I get the dw_volume_integrate term. I'm trying to integrate material parameter, where energy has been saved.
solid.update({ 'E' : get_W(stress,strain),#energy in elements }) U = problem.evaluate('dw_volume_integrate.i1.Omega(solid.E, v)',)
Or may be I should use some new variable and initialize it in post-proc. I'm asking for help in correct formulation of this integral. Should I define energy as material parameter or some new variable? And how I can do it.
Thanks in advance,
On 08/19/2013 12:51 PM, Anastasia Konovalova wrote:
Hello, sfepy group,
I have a question about post-process calculations. For my problem I used the modified examples linear_elastic.py and hyperelastic.py. After the calculation of strain and stress tensors I need to get energy of deformation
\int_{V} S : C, where C = 2*E+I.
I think I need to calculate the multiplication of tensors first, as some material variable, and then integrate it.
Yes, that should work. IIRC you can compute the energy directly as \int_{V} S:E (S is the second Piola–Kirchhoff stress tensor).
So I have 2 questions:
- How can I calculate the double scalar multiplication of two arrays strain and stress that have shape (number of elements, 1, 6, 1)? (Double scalar multiplication of A and B is a scalar that can be got as A_{ij}B_{ji}). Because of the shape of arrays I can't use numpy.inner(stress,strain). I made some function for these calculation, but perhaps, the simplier way exsists.
The six components correspond to 11, 22, 33, 12, 13, 23 components of the symmetric tensors. As the out-of-diagonal (12, 13, 23) strain components are doubled, the inner product over that axis should directly give the energy.
- Then I get the multiplication - the scalar function of energy I should integrate it. For this I get the dw_volume_integrate term. I'm trying to integrate material parameter, where energy has been saved.
solid.update({ 'E' : get_W(stress,strain),#energy in elements }) U = problem.evaluate('dw_volume_integrate.i1.Omega(solid.E, v)',)
Try using ev_integrate_mat term. The above would not work (see the terms definitions).
Or may be I should use some new variable and initialize it in post-proc. I'm asking for help in correct formulation of this integral. Should I define energy as material parameter or some new variable? And how I can do it.
As it is computed in the quadrature points, the correct way (and the easiest as well) is to use the material parameter approach. Using a variable would be possible as well, but that would involve unnecessary averaging/projection of the values from quadrature points to the FE nodes.
Cheers, r.
Well, the term works, but results are strange. Let me ask you again :)
Initially, material parameter solid.E given as 0
materials = { 'solid' : ({ ... 'E' : 0e0, # energy density },),}
In post-process, If I save the material parameter solid.E after update in vtk-file, I can see that it's changed. And values are right.
solid.update({ 'E' : get_Wh(strain,mu),#energy in elements }) W = solid['E'] out['W'] = Struct(name='output_data', mode='cell', data=W, dofs=None)
Then I try to integrate it. For the secound argument I write the unknown field u, as in some sfepy examples ( why we need some other argument except material there?). And get the null as a result.
U = problem.evaluate('ev_integrate_mat.i1.Omega(solid.E, u)',mode='eval',) print "U", U
If I put some over constant, the result will be: constant*(Omega volume). It seems, that ev_integrate_mat doesn't see the material update.
Thanks in advance
On Thu, 22 Aug 2013, Anastasia Konovalova wrote:
Well, the term works, but results are strange. Let me ask you again :)
Yeah, I thought it was too easy ;)
Initially, material parameter solid.E given as 0
� materials = { ��� 'solid' : ({ �������� ... ������� 'E' : 0e0, # energy density },),}
In post-process, If I save the material parameter solid.E after update in vtk-file, I can see that it's changed. And values are right.
solid.update({ ������� 'E' : get_Wh(strain,mu),#energy in elements ��� }) W = solid['E'] out['W'] = Struct(name='output_data', ������������������������������� mode='cell', data=W, dofs=None)
So you are using something like (material_nonlinearity.py example):
mu = pb.evaluate('ev_integrate_mat.2.Omega(nonlinear.mu, u)',
mode='el_avg', copy_materials=False, verbose=False)
out['mu'] = Struct(name='mu', mode='cell', data=mu, dofs=None)and you see the correct values, or zeros?
Then I try to integrate it. For the secound argument I write the unknown field u, as in some sfepy examples ( why we need some other argument except material there?). And get the null as a result.
The second argument is there to provide the reference element mappings etc. for the integration. Material parameters are just functions of space and time, they do not know how to integrate over elements, unlike fields.
U = problem.evaluate('ev_integrate_mat.i1.Omega(solid.E, u)',mode='eval',) print "U", U
If I put some over constant, the result will be: constant*(Omega volume). It seems, that ev_integrate_mat doesn't see the material update.
Probably. Could you send the complete file (+ a small mesh if needed) so that I can run it and look at it?
r.
So you are using something like (material_nonlinearity.py example):
mu = pb.evaluate('ev_integrate_mat.2.Omega(nonlinear.mu, u)', mode='el_avg', copy_materials=False, verbose=False) out['mu'] = Struct(name='mu', mode='cell', data=mu, dofs=None)and you see the correct values, or zeros?
Yes, something like this, and I see zeros.
Probably. Could you send the complete file (+ a small mesh if needed) so that I can run it and look at it?
r.
Here you are Thanks
On Thu, 22 Aug 2013, Anastasia Konovalova wrote:
So you are using something like (material_nonlinearity.py example): � � �mu = pb.evaluate('ev_integrate_mat.2.Omega(nonlinear.mu, u)', � � � � � � � � � � � mode='el_avg', copy_materials=False, verbose=False) � � �out['mu'] = Struct(name='mu', mode='cell', data=mu, dofs=None) and you see the correct values, or zeros?Yes, something like this, and I see zeros.
You need to use el_avg evaluation mode, when saving to an output file. �
Probably. Could you send the complete file (+ a small mesh if needed) so that I can run it and look at it? r.Here you are Thanks
The first problem I see is that get_Wh(strain,mu) returns nans in the first time step (for zero strain) instead of zeros. Could you fix that?
The material update is not that easy, I will post a solution later.
r.
I see. I'll try to fix it.
On Thu, 22 Aug 2013, Anastasia Konovalova wrote:
�I see. I'll try to fix it.
Then try using the following:
strain_qp = ev('dw_tl_he_neohook.i1.Omega( solid.mu, v, u )',
mode='qp', term_mode='strain')
solid_def = materials['solid'][0]
K, mu, kappa = solid_def['K'], solid_def['mu'], solid_def['kappa']
W = get_Wh(strain_qp, mu) #energy in elements
W = strain_qp[:, :, 0:1, :].copy() # W should have this shape! Remove when # fixed.
solid = problem.get_materials()['solid']
datas = solid.datas[solid.get_keys('Omega')[0]][0]
print W.shape, datas['E'].shape, strain_qp.shape
datas['E'] = W
Uh = ev('ev_integrate_mat.i1.Omega(solid.E, u)',mode='eval',
copy_materials=False)
print "Uh", Uh
WW = ev('ev_integrate_mat.i1.Omega(solid.E, u)',mode='el_avg',
copy_materials=False)
out['W'] = Struct(name='output_data',
mode='cell', data=WW, dofs=None)Notes:
The strain for W computation has to be evaluated in quadrature points, not averaged in elements -> 'qp' mode in the strain_qp evaluation. For some reason, this mode was not available, so I have just implemented (get the latest git version).
Instead of W I used a strain component - remove that line after the function works.
The setting of E to "datas" directly is fragile - a single element group in the mesh is supposed here.
After setting the E to the material, you have to use copy_materials=False in evaluation, so that the new value is not overwritten back with the zeros.
Let us know how it went... :)
r.
четверг, 22 августа 2013 г., 21:16:49 UTC+6 пользователь Robert Cimrman написал:
On Thu, 22 Aug 2013, Anastasia Konovalova wrote:
Let us know how it went... :)
r.
I've tested this in hyperelastic and linear elastic examples - it works.
Thank you, Robert!
On 08/27/2013 09:44 AM, Anastasia Konovalova wrote:
четверг, 22 августа 2013 г., 21:16:49 UTC+6 пользователь Robert Cimrman написал:
On Thu, 22 Aug 2013, Anastasia Konovalova wrote:
Let us know how it went... :)
r.
I've tested this in hyperelastic and linear elastic examples - it works. Thank you, Robert!
ok, good!
r.
Hello Robert! I got a question about the energy calculation again :)
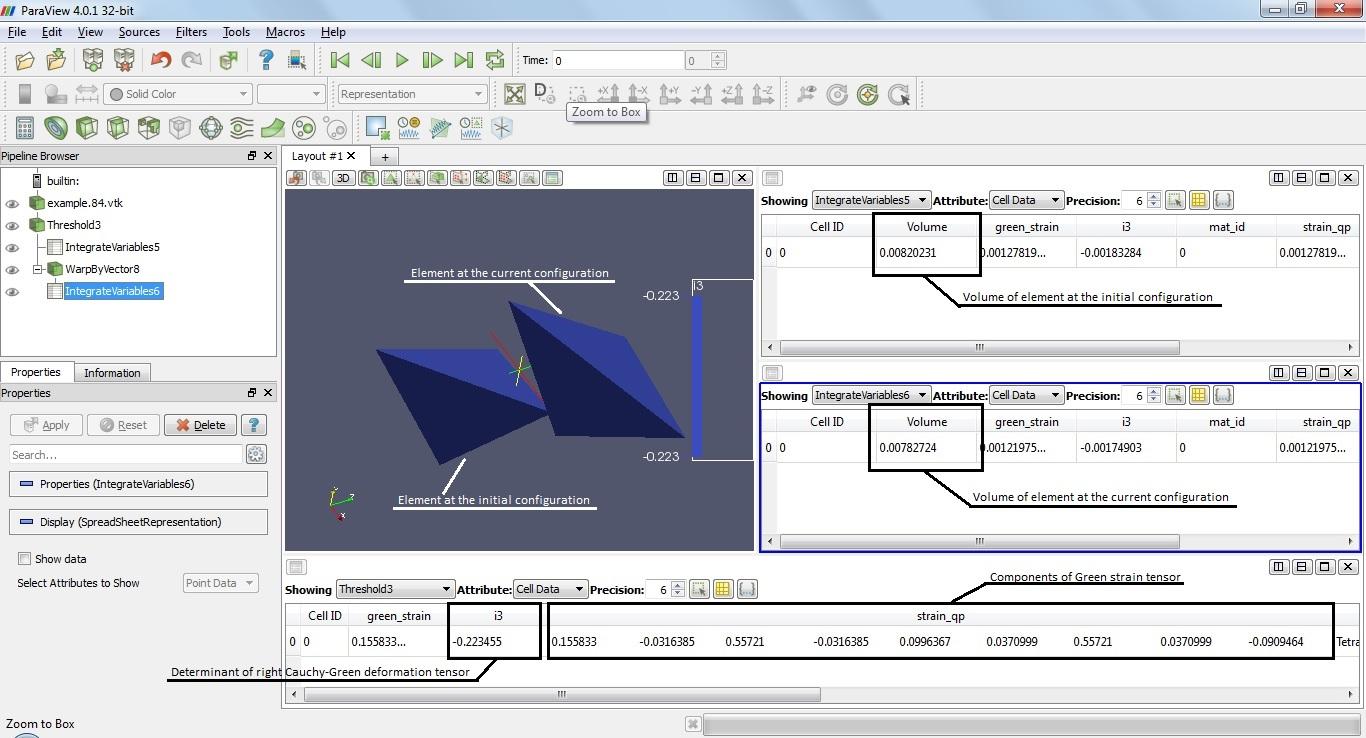
If you remember, in my programm there is function get_Wh that is called in postproc. This function compute the determinant of the right Cauchy-Green deformation tensor. When I was trying to do a large deformation, a situation arose, where in some cells the determinant is very small or even negative.
I can analyze the results on these elements in Paraview, so I see that the field of displacements of these cells are normal. I can deform my sample by displacement, so I find out that the volume of element is normal too, determinant haven't been far from 1. But If I calculate the determinant from the components of strain tensor by hands, I'll get this negative value, so that the computation of the determinant is right too.
It seems to me that this is the problem of the Green strain tensor computation.
I attached the screenshot from Paraview, where you can see the determinant value, the volume of element, and components of the Green strain tensor, the file of programm and a simple mesh, and file with the results (for paraview or some other similar programm) of one of the first steps, when the negative determinant appears.
At the function get_Wh I get the the Green strain tensor, compute the right Cauchy-Green deformation tensor (C = 2E+I), and get i3 = nm.linalg.det(C). I need the sqrt of i3 to get a J = dV/dV_0, and the error appears there.
Perhaps, it's too difficult to understand all this, so I will be glad to any advices. Thank you anyway.
{kind=link}
Hi Anastasia,
great job, you have just found a very old bug!
The following code (to be added in stress_strain() after i3 computation) should not raise errors but it does:
# black magic here...
cache = state.variables['u'].evaluate_cache['tl_common'][0][('__v_o1', 0, 0, 'volume', None)]
from sfepy.linalg import dot_sequences as dot
from sfepy.mechanics.tensors import get_full_indices
c = dot(cache.mtx_f, cache.mtx_f, 'ATB')
e = 0.5 * (c - nm.eye(3))
ii = get_full_indices(3)
c2 = cache.sym_c[..., ii, 0]
e2 = cache.green_strain[..., ii, 0]
assert(nm.allclose(nm.sqrt(i3), cache.det_f, rtol=1e-15, atol=1e-15))
assert(nm.allclose(c, c2, rtol=1e-15, atol=1e-15))
assert(nm.allclose(e, e2, rtol=1e-15, atol=1e-15))The last line assertion fails, so the Green strain e2 as computed by the dw_tl_he_neohook term call differs from e computed by definition (0.5 * (F^T F
- I)).
It has escaped me for so long as we have used the green strain only in postprocessing to make "colorful images"... The Green strain has not been used in any further computations. The stresses are ok - they are computed from the C invariants directly.
The problem (doubled out-of diagonal entries of E) is now fixed in the repo and your example gives positive i3 in all time steps.
Thanks again for noticing the problem!
Cheers, r.
PS: If you would like to have a term/terms for all the attributes of the evaluate cache above (do 'print cache' to see them), add an issue, please.
On 11/06/2013 08:45 AM, Anastasia Konovalova wrote:
Hello Robert! I got a question about the energy calculation again :)
If you remember, in my programm there is function get_Wh that is called in postproc. This function compute the determinant of the right Cauchy-Green deformation tensor. When I was trying to do a large deformation, a situation arose, where in some cells the determinant is very small or even negative.
I can analyze the results on these elements in Paraview, so I see that the field of displacements of these cells are normal. I can deform my sample by displacement, so I find out that the volume of element is normal too, determinant haven't been far from 1. But If I calculate the determinant from the components of strain tensor by hands, I'll get this negative value, so that the computation of the determinant is right too.
It seems to me that this is the problem of the Green strain tensor computation.
I attached the screenshot from Paraview, where you can see the determinant value, the volume of element, and components of the Green strain tensor, the file of programm and a simple mesh, and file with the results (for paraview or some other similar programm) of one of the first steps, when the negative determinant appears.
At the function get_Wh I get the the Green strain tensor, compute the right Cauchy-Green deformation tensor (C = 2E+I), and get i3 = nm.linalg.det(C). I need the sqrt of i3 to get a J = dV/dV_0, and the error appears there.
Perhaps, it's too difficult to understand all this, so I will be glad to any advices. Thank you anyway.
Thanks for the answer, Robert! And excuse me for the long response.
I'm not sure, that I understand you clearly. I have added changes from Git, and the computation of determinant in my programm is correct now. And your "black magic" code don't produce error any more. So you kill this old bag, right?) And strain can be used for futher computations now? Thank you very much!
As for the terms and issues, I compute determinant and other values for the energy calculation. If it is possible to make terms for the energy density function for hyperelastic material, it would be great and I'll add the issue as soon as I can:)
четверг, 7 ноября 2013 г., 16:34:59 UTC+6 пользователь Robert Cimrman написал:
participants (2)
-
 Anastasia Konovalova
Anastasia Konovalova -
 Robert Cimrman
Robert Cimrman